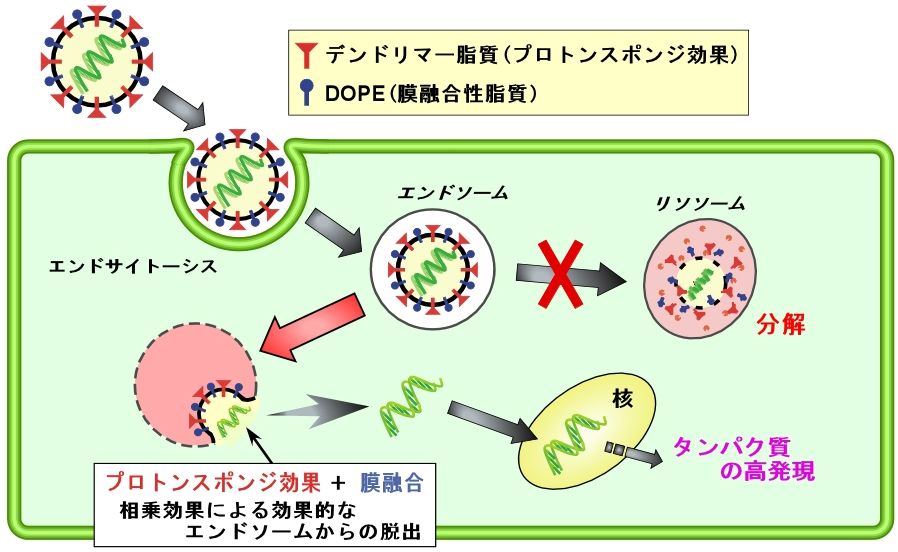
遺伝子操作の概要
-
遺伝子の機能を調べるにはいくつかのテクニックが必要である。
生物体内(in vivo)における特定の遺伝子はいくつかのコピーが存在するものの、その遺伝子が何を意味しているのか、発現するとどうなるのか、変異が起こればどうなるのかを調べることは困難である。
したがって、その遺伝子のみを取り出して、遺伝子の特性を生物体外(in vitro)で調べる必要がある。
それらの過程には
クローニング:扱いやすくするために対象遺伝子を担うDNAの数を増やす
シークエンシング:遺伝子の配列を読む
過剰発現:遺伝子をタンパク質に翻訳し、その機能を理解する
の三段階を経る。
クローニングや発現の前には、サブクローニングや発現ベクターへの遺伝子の導入といったプロセスを経ることもある。
クローニング
-
クローニングとは、遺伝子のクローンを作成する実験である。
遺伝子のクローンを作成するにはある程度の配列がわかっていることを前提に現在2つの方法が実用化されている。
ゲノムDNAを制限酵素で切断し、サザンブロッティングで目的遺伝子を含む配列を同定する方法 PCR法を用いて目的の遺伝子配列を増幅する方法 遺伝子配列がわからない場合には、目的のタンパク質を対象生物内から精製し、タンパク質N末端配列を決定した後、ミックスプライマー(アミノ酸とその遺伝暗号に対応するパターン全てを含む複数種のプライマー、詳しくは当該記事にて)を用いてクローニングを行なうことができる。
上記いずれのケースにおいても、単一のDNA配列のみを増幅した、あるいは精製したのみではヌクレアーゼによって分解を受けてしまう。
したがって、目的DNA配列をクローニングベクターに導入し、大腸菌を用いてベクターを増幅することを含めてクローニングという実験が完結する。
なお、真正細菌は遺伝子に介在配列を持たないためにDNAから遺伝子をクローニングすることが可能だが、真核生物の場合はイントロンをのぞいたエクソン部分のみを抽出する必要がある。
これはスプライシング後のmRNAを精製し、逆転写PCR (RT-PCR) を行なうことによってクローニングが可能となる。
遺伝子研究の応用
-
遺伝子導入とは、上記にあげた遺伝子の実験系を用いて目的遺伝子の宿主でない他生物にクローニングしたベクターを導入し、その遺伝子が有効な形質を発現できるように仕向けることである。
例えば、特定の除草剤に対して耐性を持つような作物や、霜害を防ぐ糖タンパクを生産できる作物(アイスマイナス)などはその一例である。
しかしながら、導入したベクターが花粉などを通じて拡散し、除草剤耐性を持っていなかった雑草にまでそうした形質が導入される危険性を指摘され、このような遺伝子組み換え実験は厳しく規制された状況である。
遺伝子組み換え実験による物理的規制は
P1:一般的な実験室
P2:病原性を扱うような実験室
P3:バイオクリーンルーム(室内に流入する空気は全てHEPAフィルタ(0.22μm方形のフィルター)を通している)
P4:遺伝子組み換え対象生物に触れずに実験できる実験室
というランクが付けられており、危険な遺伝子組み換え実験を行なう場合にはそれ相応の規制のランクの敷かれた実験室で行なうように義務付けられている。